


Broken Bioweapon
Lack of mRNA Integrity in Pfizer Batches: All Regulators Knew This When they "Pretend-Approved" the Shots

Background:
Lack of mRNA integrity and product impurities (fragmented nucleic acid chains) were found in Pfizer’s product days before it was authorized for market. mRNA integrity, and conversely, its instability, is one of the most important variables relevant to all mRNA vaccines. Pfizer and BioNTech repeatedly stated that the efficacy of the product is highly dependent on the quantity of the sufficiently intact mRNA molecule. Even a minor degradation reaction, anywhere along a mRNA strand, can severely slow or stop proper translation performance of that strand and thus result in the incomplete expression of the target antigen.
I reported on this issue about a year ago, but since I have larger audience now I am re-publishing my analysis here. The integrity of key active ingredient – mRNA molecule as measured by the %mRNA integrity and % of fragments (Late Migrating Species, LMS) in each manufactured batch is highly and unacceptably variable. This was identified by the regulatory reviewers at EMA and FDA. The discussions around this issue are recorded in numerous documents that were released from EMA, at the end of November 2020, including email exchanges between EMA staff and senior executives. The “solution” to this issue was to lower the acceptance standard for the %mRNA integrity in the active substance for shelf live to just >50%. In other words, the “other” 50% of the nucleic acid chains and other impurities can be present in the active substance, do not need to be characterized (no matter the length or composition of the RNA molecules) and the product is still deemed “acceptable”.
An extremely wide variation of the integrity of the active substance in bulk material of the product and abundant presence of uncharacterized nucleic acid impurities means that batches of different formulation - and thus different potency and safety profiles - are being produced. This variation is further amplified when hundreds of liters of the bulk material is filled in small quantities into 0.45 ml vials and subsequently manually divided into doses by untrained and unsupervised staff at the vaccination centers.
Both the regulators and Pfizer to date have not disclosed the acceptable ranges or testing methods for the key ingredients of the vaccine product, neither in bulk product nor in a vial or a dose (as dispensed), and claim “commercial secrets” prevent them from doing so.
33 Commercial Batches Manufactured Prior to December 2020:
Records released by the EMA leak contain some manufacturing and testing details for 33 commercial scale batches made by Pfizer between August and late November 2020. The fact that commercial batches were manufactured prior to the regulatory review and prior to any safety or efficacy data available for the product is a violation of CFR21 - current Good Manufacturing Practices. To clarify, a company can manufacture a volume of product prior to approval, but once the regulatory review finds significant problems in the submission (as was found here) - shipping these batches commercially without demonstrating that whatever issues were found did not affect the product safety and efficacy is a big problem. Given the number of significant and major objections and the short span of time (about 2 weeks before product was shipped) it is not possible that these problems were resolved. That’s why no sane and well-intentioned manufacturer would ever do such a thing - produce millions of doses of expensive product “at risk” before finding whether it is actually safe. However, geniuses at BARDA believe this is hip, cool and “innovative” and only the losers in the private sector should follow laws and regulations. When the government is in the business of “saving lives”, no laws apply and nobody is concerned with boring regulations:

Back to the 33 “pre-authorization” batches:
Specifically, these batches and all information used in my analysis were listed in the document titled: “COVID-19 Vaccine (BNT162, PF-07302048) BB-IND 19736 Response to CBER Comments Received on 20 November 2020 Regarding Overall CMC Information.” The document is dated November 25, 2020. It was part of the documents that were leaked from the EMA. Here’s the title page:
At the time of issue of this document, the manufacturing facilities were deemed not in acceptable compliance with the current Good Manufacturing Practices by the regulatory authorities (both EMA and FDA). CGMP a set of regulatory requirements guiding quality, purity, stability and reproducibility and labeling of mass-produced drugs and vaccines. The cGMP regulations for drugs contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing of a drug product. The regulations make sure that a product is safe for use, and that it has the ingredients and strength it claims to have.
The non-compliance with cGMP was recorded as Major Objection #1 by the EMA reviewers, and in addition, the issue of lack of mRNA integrity and presence of significant uncharacterized impurities in the form of nucleic acid chains (LMS) was raised and recorded as Major Objection #2. Here are the slides from EMA-BioNTech meeting discussing the “comparability” issue, i.e., the batches used to manufacture clinical trial material (using Process 1) were very substantially different from the batches manufactured at commercial scale (Process 2) and there was no way to reconcile this without re-running a clinical trial. But of course that’s not what they decided to do. The response was to simply lower the acceptance standard, which was arbitrarily set to begin with, from >70% to >50% of the batch would need to conform to the specification for it’s key active ingredient - an intact, full length mRNA molecule of certain specification.

The regulators and manufacturer knew there were going to be “truncated” molecules, and had no idea what mutant proteins that would create…

They had the tests showing significant profile differences between Process 1 and Process 2 batches (note the “bumps” before the main spike)…

The %mRNA integrity was not comparable between Process 1 and Process 2 (and strictly speaking, within each process, too)…
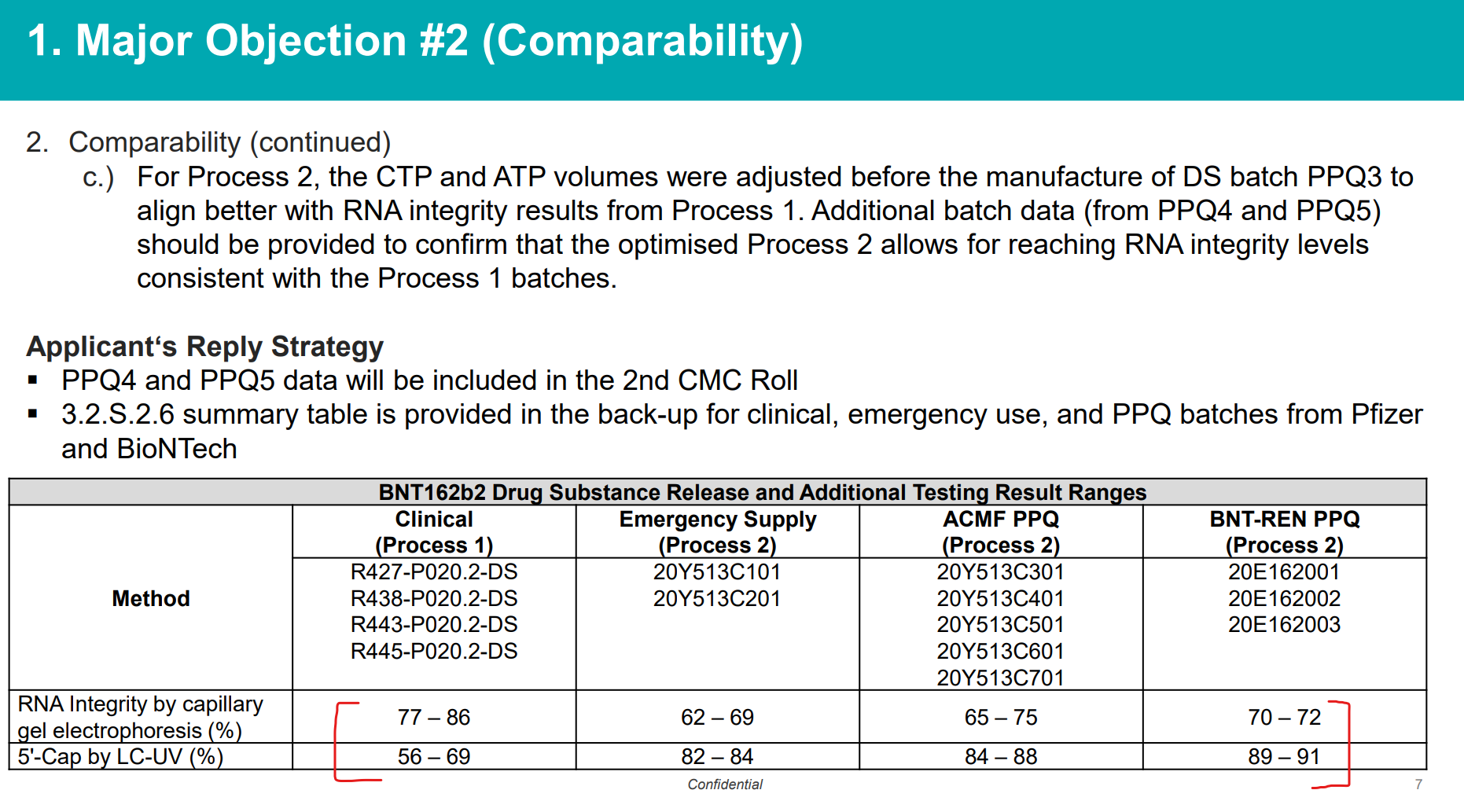
The “Applicant’s Reply Strategy” was to simply lower acceptance threshold without any data showing that any of this is ok…
It was clear that the mRNA breaks and degrades during production (drug substance manufactured into drug product), and subsequently during shelf storage. Of course this process continues during transportation and manual dose preparation, because those “moving at the speed of $ience! (TM)” are not concerned with any of this pedestrian stuff…

To summarize, in its rolling review in November 2020, the EMA noted that a decrease in RNA integrity, which is a critical quality attribute, was observed such that RNA integrity of Process 1 (78.1-82.8%) were much higher than the commercial scaled up Process 2 (59.7%). Furthermore, it was not possible to determine if the differences in RNA integrity were qualitative, quantitative, or both. Truncated mRNAs in Process 2 were also not defined. At the time of the Conditional Marketing Approval in EU it was not known what proteins were coded for, if any, and their clinical effect was not known. It could not be excluded that proteins different from the intact full-length spike would be expressed. It is also possible that truncated mRNA may affect translational efficacy or modify the immunostimulatory profile. Even if these truncated mRNA are not translated, they are considered impurities and should minimized or eliminated as much as possible so that the same amount of intact pure mRNA is present in each batch.
There were additional numerous objections and concerns of the regulator (100+ of formal regulatory objections were found in the EMA documents from late November 2020).
The 33 Pfizer batches contained a total of approximately 6 million vials for approximately 29 million vaccine doses. Note that at the time of approval, Pfizer multi-dose vials were for 5 doses vs 6 specified on the label today. These batches were shipped commercially to locations in the US and Europe (and possibly other regions) despite the documented noncompliant manufacturing, or serious regulatory concerns. Of these 33 batches, 26 batches have adverse event reports associated with them in VAERS database.
The table below provides some details about these batches as well as the numbers of adverse events and deaths found in VAERS. It should be noted that since these batches were distributed to many regions, including US, Europe and other locations, parts of the batches or whole batches may have been sent to locations that did not submit adverse event reports to VAERS system. In addition, VAERS system is known to under-represent the true adverse event rate by 40-100 times. Therefore, the true rate of the adverse events associated with these batches is likely to be several times greater than what is shown here.
Finally, all these batches below have listed expiration date as 12/31/2069 (!) - sheer nonsense.
Table 1.

EMA and FDA documents contained some detailed batch analyses for 18 batches, including the %mRNA integrity metric. I compared this parameter to deaths, permanent disabilities, and total adverse events (including non-serious) reported for the batch number in the CDC Vaccine Adverse Events Reporting database (VAERS). The regression analysis demonstrated significant statistical relationship between %mRNA integrity and deaths and permanent disabilities reported per batch. The deaths, disabilities and total adverse even rates were adjusted for the number of vials produced in a batch. Only a mild inverse relationship was found for total adverse events that included non-serious events. Figures 1-3 summarize the results.
Figure 1.
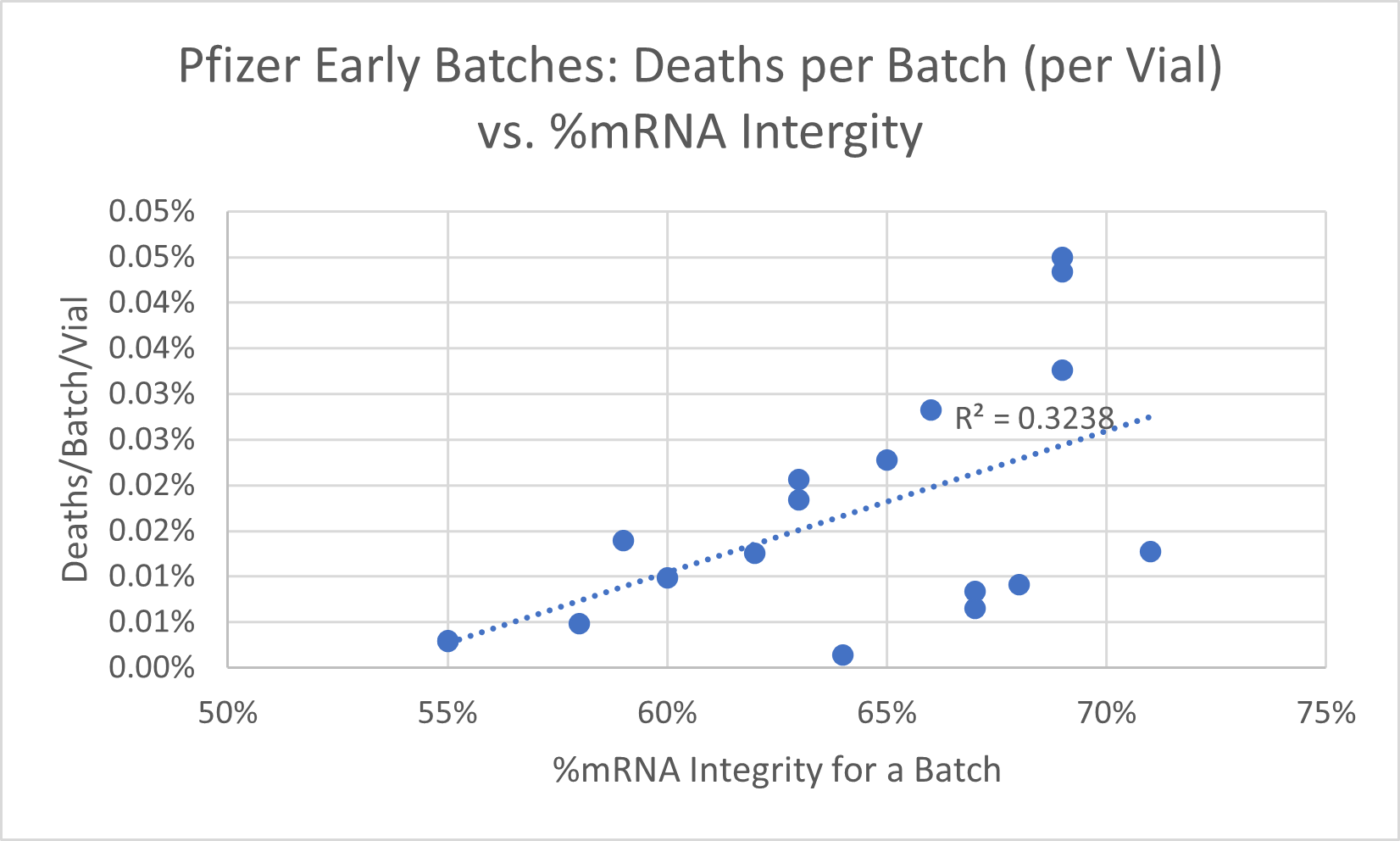
Figure 1 demonstrates significant positive slope (R2 = 0.33) between %mRNA integrity and death reported per batch, adjusted for the number of vials produced – higher integrity in the early batches leads to more lethal events reported for the batch in VAERS. This indicates that, at least this group of batches, was a deadly (death causing) product when manufactured to higher conformance to the specification.
Figure 2.

Figure 2 demonstrates the significant inverse association (R2 = 0.37) between %mRNA integrity and permanent disabilities reported per batch, adjusted for the number of vials produced – higher integrity in the early batches leads to relatively fewer permanent disability events reported for the batch in VAERS. It is possible that the deadly product wasn’t as potent in some of the vials in a batch and didn’t kill but disabled people. Also possible that it killed older/more fragile people and disabled younger ones (this was not specifically tested in my analysis).
Figure 3.

Figure 3 demonstrates that only a mild inverse trend was found when total adverse events reported per batch vs. %mRNA integrity metric are examined. Total includes all serious and non-serious reports and deaths together.
Discussion:
At the time of Conditional Marketing Authorization in the EU, it appears Pfizer’s product would not normally meet the most basic quality controls expected of an injectable pharmaceutical. The basic pharmacology was unknown, the quantity of produced spike was unknown, and the quality and reproducibility of the production of the commercial batches were extremely poor.
My dataset exhibits alarming trends – extremely large ranges of adverse events, deaths, and disabilities as well as strong statistical association between %mRNA integrity metric with deaths/ permanent disabilities. mRNA integrity is only one of dozens of critical quality attributes of this product. The manufacturer has never made the batches available for an independent testing and characterization, sequencing, or assessment of the mRNA or other nucleic acid fragments and impurities present in them. Thus, millions of people injected with these substances can never know what exact sequence(s) of nucleic acids were present in these injections, and what proteins may be expressed and what health impact they may have. Sadly, at least 1000 people lost their lives to these “unknown” effects by a poorly made product shrouded in “commercial secrecy”. Without the knowledge of exact nature of the composition of various batches it is not possible to make definitive conclusions or offer good treatment options to the survivors of these injections.
Pfizer product does not meet the robust quality requirements of a pharmaceutical grade product given to billions of humans and contain large amounts of impurities. The manufacturer and regulators actively avoid transparency, disclosure, or correction of these issues. These acts can no longer be considered “mistakes” under “rush” conditions. The products must be stopped and recalled worldwide and thorough investigations must be initiated.
Art piece for today: Sunflower, oil on panel, 18x24 inches.

I am a pharmacist. I was immediately concerned about the integrity of the mRNA and the human ability to preserve it in transport and storage. I still do not understand why more pharmacists weren't/aren't also skeptical. I feel like a lone wolf in my profession.
Layman understanding. The purer the MRNA the more severe adverse events.