


My research summarized for a book chapter.
I have been asked to contribute a book chapter by several authors. Hope this will be published one day.

Non-Enforcement of Pharmaceutical Law Revealed Intent to Harm
In early 2020, I grew increasingly alarmed by the government’s nonsensical and counterproductive actions marketed as “covid response”. The realization that something truly fraudulent was occurring came to me around spring-summer of 2020 when the suppression of hydroxychloroquine and other early effective covid treatments by the government became obvious.
The HHS (CDC & FDA) went on massive social media campaigns demonizing generic drugs such as hydroxychloroquine and ivermectin, calling them “fish tank cleaner” and “horse dewormer” - which was simply shocking to me as I have never witnessed the health authorities lying so openly and brazenly. The CDC’s own research, published in 2005, concluded, “Chloroquine is a potent inhibitor of SARS coronavirus infection and spread”[1]. The U.S. had stockpiled hydroxychloroquine for exactly this sort of emergency.
The HHS and state agencies and their “fixers” in academia and mass media were promoting outright lies about this well-characterized generic medicine with a long history of safe use. A turning point for me was when a study by a US tech company called Surgisphere[2] claiming to have collected all data for all covid patients at the time (tens of thousands in all countries of the world) was published in Lancet claiming that hydroxychloroquine caused numerous cases of lethal arrhythmia. The group claimed to have compiled an enormous dataset from all over the world in a matter of weeks! Sure enough, in about 14 days Lancet retracted the study as it was fraudulent, and no such dataset ever existed. But by then the mainstream media ran the fake headlines which were never retracted. I decided to investigate the data for myself, and turned to the only dataset that was available to me - Vaccine Adverse Event Reporting System (VAERS)[3].
Vaccines in general are regulated as pharmaceutical products, meaning that they have a requirement to comply with the current Good Manufacturing Practices[4] – a set of regulatory laws governing quality control, safety, and accurate labeling of pharmaceuticals. A consumer or a healthcare provider usually cannot detect (through smell, touch, or sight) whether a drug product is safe or if it will work, and therefore safety testing and assurance processes are needed. This formal system of controls at a pharmaceutical company, if adequately put into practice, helps to prevent instances of contamination, mix-ups, deviations, failures, and errors. More importantly, if the process of manufacture of a substance cannot be assured, that substance cannot exist as a pharmaceutical – reliable, predictable, and controllable by precise dosage. If a company is not complying with cGMP regulations, any drug it makes is considered “adulterated” under the law - a de facto potential poison, whether purposeful or accidental. Most product recalls are voluntary by the manufacturers; however, the FDA has power to enforce cGMP compliance and force recalls, product seizure and a variety of other regulatory actions to ensure compliance.
I needed a historic comparator, and I decided to study the patterns of VAERS data for Covid 19 injections in comparison to the historical flu vaccines from a variety of manufacturers. I compared the covid injections to flu vaccines in terms of the total numbers of serious adverse events and deaths and, in addition, looked at how many we reported per each lot number. It was an important question, as from my experience in the pharma industry, this analysis would be an important indicator of manufacturing quality control. I later met and collaborated with a group of citizen analysts around this topic, including Dr. Mike Yeadon, a former senior R&D executive from Pfizer and some of the earliest, and Craig Paardekooper, a British pharmacy student. Craig made many analytical discoveries in VAERS lot data and started the “How Bad is Your Batch” website dedicated to this issue[5]. The website became an invaluable resource for all questions related to injection batches and by the end of 2022 received over 100 million visitors from all over the world. We called our group “Team Enigma” in honor of the analysts that cracked the Nazi code during World War II. I started video recording presentations of our data and posting them on alternative media channels as mainstream social media was in full-on suppression mode.[6]
I queried VAERS data many times over 2021-2022 using historical data from seasonal flu vaccines lot-to-lot as a comparator. Right away it was obvious that the difference was astounding: covid injections had 15-41 times more serious adverse events and 40-152 times more deaths per lot. Even more troubling was the incredibly high lot-to-lot variation for covid shots compared to the flu vaccines, even after adjusting for the lot sizes in doses. It is expected that for a product manufactured in compliance with the cGMP all manufacturing lots will have very similar rates of adverse events, very close to zero, and that held largely true for the flu vaccine data. In contrast, the covid shots varied roughly 1000% lot-to-lot. Some lots had very few reports, and some had 5000-6000 reports, including 1,000-2,000 serious reports and hundreds of deaths. The variability was not explained by the sizes of lots, nor by any normal demographic data adjustments such as age or gender of the recipients. It was clear that the manufacture was recklessly uncontrolled. When a pharmaceutical product is made without adherence to the manufacturing quality regulations, any product thus produced is a potential poison, and is deemed as such by law – a de-facto adulterated product[7].
Over time direct evidence of non-compliance with cGMP became available, validating my earlier conclusions. Evidence included the European Medicines Agency (EMA) “rolling review” of Pfizer’s Chemistry and Manufacturing Controls (CMC) documentation. The discussions around this issue are recorded in numerous documents that were leaked from EMA at the end of November 2020, including email exchanges between EMA staff and management. Authenticity of the documents and emails was independently verified by the British Medical Journal.[8]
The EMA reviewers noted lack of cGMP compliance as “Major Objection #1,” in addition to more than 100 additional Major Objections and Critical Observations. Yet, all regulatory objections were disregarded, and the product went on the market in Europe and worldwide shortly after.
Despite obvious safety signals including Pfizer’s own pharmacovigilance report showing 1200+ deaths[9] within first 2 month of mass vaccinations, thousands of death reports in VAERS, and millions of reported injuries in other government tracked databases like V-Safe[10], there were no recalls or investigations by the manufacturers or health agencies. Not a single health official “noticed” it, and furthermore, when questioned, mounted a wall of denial in response.
Historically, a handful of reports of deaths associated with the use of the product is all that’s needed to initiate a pharmacovigilance action – a recall of a batch of product, investigation, or an entire product recall – all these measures are available and have been successfully used in a variety of product contamination and adulteration cases[11]. After thousands of deaths are recorded with the use of a product and no action is taken by anyone – manufacturer, regulators or law enforcement authorities, the situation should be considered intentional. The flat refusal to acknowledge deaths and injuries by the government officials and pharma companies signaled the intent. The intent to harm.
The “Pseudo-Laws”: EUA Countermeasures Under Public Health Emergency.
In mid-2022 I ran across a brilliant analysis titled “American Domestic Bioterrorism” posted online by my now friend and colleague Katherine Watt, a paralegal and an independent journalist researching US law history.[1] Her writing provided the critical piece of the puzzle I was looking for: it explained how the criminal cartel, including actors in the US government, military, and private companies “legalized” and implemented the covid atrocity.
Katherine found a crucial provision in US law: 21 USC 360bbb-3(k) which stated that use of EUA-covered medical countermeasure (MCM) products including masks, diagnostic tests, bioagent injections, and other drugs, devices and biologics, once so classified by the HHS Secretary and his/her delegees, “shall not be considered to constitute a clinical investigation.”
This provision and the entire structure of the EUA Countermeasures law uncovered by Katherine revealed how the US Government systematically removed all consumer safeguards for the products designated as military “countermeasures” under contrived, arbitrary, self-proclaimed conditions of “public health emergency”, and then lied to the public pretending that these were properly approved safe and effective pharmaceuticals.
Under federal law, FDA must approve any new drug product prior to a manufacturer introducing it into interstate commerce.[2] This process requires manufacturer to open an Investigational New Drug (IND) application and obtain an exemption from the FDA for its use in regulated clinical research (trials). This regulated process is therefore referred to as an “investigational” regulatory pathway. It requires a manufacturer to conduct regulated clinical research (trials) under the IND, obtaining Institutional Review Board’s (IRB) approval for clinical trial protocols, independent safety monitoring oversight, and informed consent from clinical trial volunteers. In addition, manufacture of the drugs and biologics subject to the investigational status is regulated by the current Good Manufacturing practices (cGMP)[3]
EUA Medical Countermeasures are a radically different, non-investigational drugs, biologics and devices deployed under FDA’s authorization power known as the “Emergency Use Authorization” (EUA) process[4]. The EUA process is used only when the United States Secretary of Health and Human Services declares an emergency[5] and the EUA law covers only instances of an attack with a chemical, biological, radiological, or nuclear (CBRN) agent(s). The EUA law does not anticipate issuing EUAs for products to treat or prevent viral or bacterial outbreaks.
By law, the EUA process is non-investigational[6]: while the manufacturers may choose and FDA may ask to undertake some of the activities typically expected from an investigational clinical trial and manufacturing validation process, none of the typical regulatory standards are applicable in an enforceable way.
FDA has the discretion to issue an EUA if the applicant shows, or HHS Secretary thinks that its product “may be effective” in treating the relevant disease or condition[7]. No other criteria for approval apply in an enforceable way. Thus, the FDA will approve EUA products on incomplete information so long as the applicant shows that the “known and potential benefit of the product” merely “outweigh[s] the known and potential risks”[8] and considers it unlikely that “comprehensive effectiveness data” will be available before an EUA grant.
In contrast, for an investigational drug (under normal regulatory approval process) the FDA “shall” deny approval if the applicant “do[es] not show that such drug is safe.”[9]
Therefore, the EUA status of an MCM precludes collection of the regulated clinical trial data and thus precludes reliable, valid scientific knowledge of risks and benefits associated with the EUA Countermeasure while it remains non-investigational.
There is no strict requirement for an investigational new drug regulation (IND), nor institutional review board (IRB) approval of a clinical trial protocol and no informed consent. This fact was revealed in an FDA Vaccines and Related Biologicals Advisory Committee meeting on October 22, 2020, where Doran Fink answered questions on behalf of the Operation Warp Speed[10]. When asked whether the vaccines under OWS were going to use Expanded Access Use (an investigational pharmaceutical pathway to approval), his answer was clear – they were not, because an investigational pathway required informed consent, something that would pose “operational complexity” for mRNA shots. Here is his exact statement from available transcript: “The differences between expanded access use and Emergency Use Authorization are that expanded access use is done -- or is carried out under FDA's investigational new drug regulations. So among many other things, those regulations require use of an institutional review board and also obtaining informed consent from recipients of the investigational vaccine according to regulations for clinical investigations -- research use of investigational vaccines. And so operationally speaking, an expanded access protocol would add some complexity, and that is why Emergency Use Authorization is being considered primarily as the mechanism for addressing the public health emergency that has been declared.”
In fact, the EUA process precludes meaningful informed consent from the recipients of the product altogether. Congress mandated that FDA directly inform health care professionals and product recipients of any “significant known and potential benefits and risks.”[11] However, given that formal regulated clinical trials are neither required nor possible for a non-investigational EUA product, there is no effective way to collect and collate reliable and scientifically valid information on risks and benefits of an EUA, thus making the informed consent mandated by Congress meaningless and unobtainable.
Furthermore, there are no required standards for quality-control in manufacturing; no inspections of manufacturing procedures; no lot-release testing and no prohibition on wide variability among lots; no prohibition on adulteration; and no required compliance with Current Good Manufacturing Practices. EUA products, even though unregulated and non-standardized, “shall not be deemed adulterated or misbranded.”[12]
Thus, I was able to confirm that covid countermeasures are exempt from normal regulations and consumer protections that apply to medical products:
· To the extent that "use" of Covid-19 products "shall not constitute clinical investigation," use of such products is authorized even if there is no safety or efficacy data, even if such products are toxic and ineffective.
· Investigators, researchers, physicians, nurses, pharmacists and other individuals involved in product dispensing, use, or administration to human beings apparently have no legal obligations to comply with laws and regulations that applied previously to use of experimental, investigational, unapproved or approved biological products or devices, including compliance with informed consent laws, medical monitoring of recipients during product use and post-administration monitoring and reporting of adverse effects.
· Recipients of such products are not legally recognized as experimental subjects or patients receiving experimental, authorized, or approved products, because "use" of the products "shall not constitute clinical investigation." There is no stopping condition, because there is no legally relevant "clinical investigation" to be stopped.
· On the basis of a self-declared "public health emergency" and self-declared classification of products as "emergency use medical countermeasures," including an unreviewable determination as to the relative risks posed by a communicable pathogen as compared to "medical countermeasure" products, the Secretary of Health and Human Services can suspend informed consent obligations and rights, on behalf of the entire American population.
· "Vaccinators" are thereby authorized by the HHS Secretary to withhold information about product ingredients; vial contents; potential individual risks and benefits based on individual health conditions; treatment alternatives; and the option to accept or refuse the products.
The FDA and other governments’ drug regulatory agencies have not withdrawn authorizations or approvals of the covid countermeasures, despite millions of documented injuries and deaths during the initial deployment phase, because the products are not legally recognized as medicines. Instead, they are weapons, biological and chemical and psychological/informational weapons of mass destruction presented to the public under the guise of “public health”.
The “Theater” of a Military Operation
While misrepresenting the covid illness as a “zoonotic viral respiratory infection”, from early 2020 the US Government organized itself for an act of war. Specifically, on March 13, 2020: “PanCAP Adapted U.S. Government COVID-19 Response Plan” (PanCAP-A) stated that United States policy in response to SARS-CoV-2 was set not by the public health agencies designated in pandemic preparedness protocols (Pandemic and All Hazards Preparedness Act,[13] PPD-44,[14] BIA), but rather by the National Security Council (NSC). NSC does not have regular attendees from public health agencies and its focus is national security, intelligence and foreign policy matters.
While the Operation Warp Speed[15] was presented to the public as a “collaborative” “all of government” effort of the DOD and HHS, the DOD was formally the Chief Operating Officer (general Gustave Perna was named as the COO), while HHS had only the Chief Science Advisor position.[16]
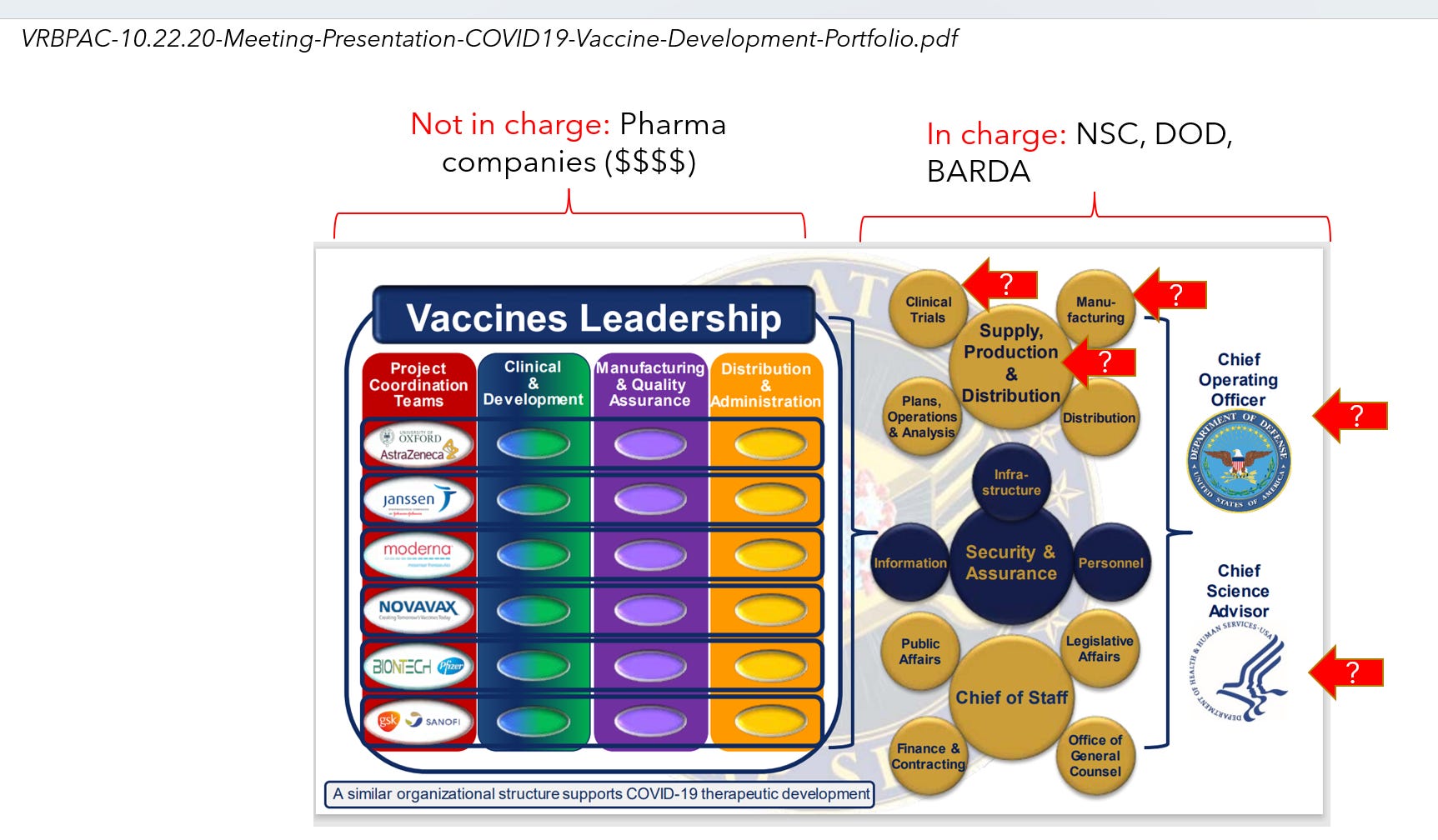
Notably, the next senior most layer of the organization was controlled by the US Government while the pharmaceutical companies were the third level down in this organization. The unclassified October 2020 documents from Operation Warp Speed presentations at the FDA's Vaccines and Related Biological Products Advisory Committee revealed control of the US Government over nearly all product design and implementation aspects of the development and clinical trials for Covid countermeasures.
On 5 March 2020, the U.S. Army’s Brigadier General Michael J. Talley announced at a press briefing that the DoD would lead a “whole of government approach” as part of its medical countermeasures response to protect “the citizens of the world”.

US has separate branches of government and separate federal agencies for many reasons. Importantly, this was envisioned by the framers as a system where federal agencies’ authorities are limited, and where different branches of government serve as checks or limits to each other. All those reasons apparently could be disregarded in an instant, under a manufactured “emergency”.
At the time of the OWS announcement I thought it was odd that there was a need to bring DOD into this activity at all. What do they know about making drugs? What do they know about pharmacy distribution chain in the US (which is fast, traceable, regulated by the states, very experienced, and is already established everywhere)? I later learned about the real role of the DOD in the “covid” exercise.
Debbie Lerman, a journalist writing for Brownstone Institute[1], revealed that one of the main reasons this “partnering” between HHS and DOD was the use of Other Transaction Authority (OTA). The OTA method of contracting allows federal agencies to order otherwise-regulated products bypassing any such regulations, as well as financial accountability mechanisms that cover standard government contracting, and other laws that regulate disclosure and Intellectual Property (IP) derived from publicly funded research.[2] Quite reasonably, different federal agencies have different scope of the OTAs. The scope of HHS’s OTA did not permit manufacturing pharmaceuticals at scale, especially not before satisfying regulatory requirements for safety. OTA was written and codified as a way for the military to acquire weapons and other necessary equipment without a lot of bureaucratic red tape. It covers research and development, prototypes, and subsequent manufacturing. The only OTA for a public health agency is for the HHS and it only covers Research & Development, not prototypes or manufacturing.
Even the R&D OTA given to the HHS still requires products to be manufactured “in a form that satisfies the regulatory requirements” for drug and vaccine safety. There was no way HHS could have used its very limited OTA to sign contracts for hundreds of millions of novel medical products.
So, what did HHS do?
Acting as separate federal agencies within the limits of their authorities, neither HHS nor DOD would have been able to order 100 million doses of unapproved, untested, previously thoroughly failed “vaccine”. They “partnered” to break the constraints of their authorities.
Another very useful insight on EUA Countermeasures law was that the utilization of EUA in this “partnering” scheme is evidence that the relevant US Government officials never believed covid was a naturally occurring viral pandemic. They knew it was a chemical, biological, radiological, or nuclear (CBRN) agent or combination of such agents. That is because the EUA law does not include naturally occurring viral outbreaks. The fact that the senior government executives lied about the “wet market zoonotic jump”, utilizing massive network of DHS and intelligence contractors to censor any social media mention to the contrary, prosecuting and smearing any credentialed person who pointed to the holes in their narrative, performing endless clown-show Congressional hearings about whether it was a zoonotic jump or a Wuhan leak, and currently continue to pretend covid was a viral pandemic only points to one thing - this attack being perpetuated by those who lie and cover it up, i.e. the US Government officials themselves.
In essence, the covid response used the pretext of a possible ‘biological attack’ to justify pre-planned “rapid response partnerships” between government and biodefense contractors in industry and academia to facilitate the international deployment of emergency medical countermeasures (MCMs), allegedly to protect American civilians and “the citizens of the world”.
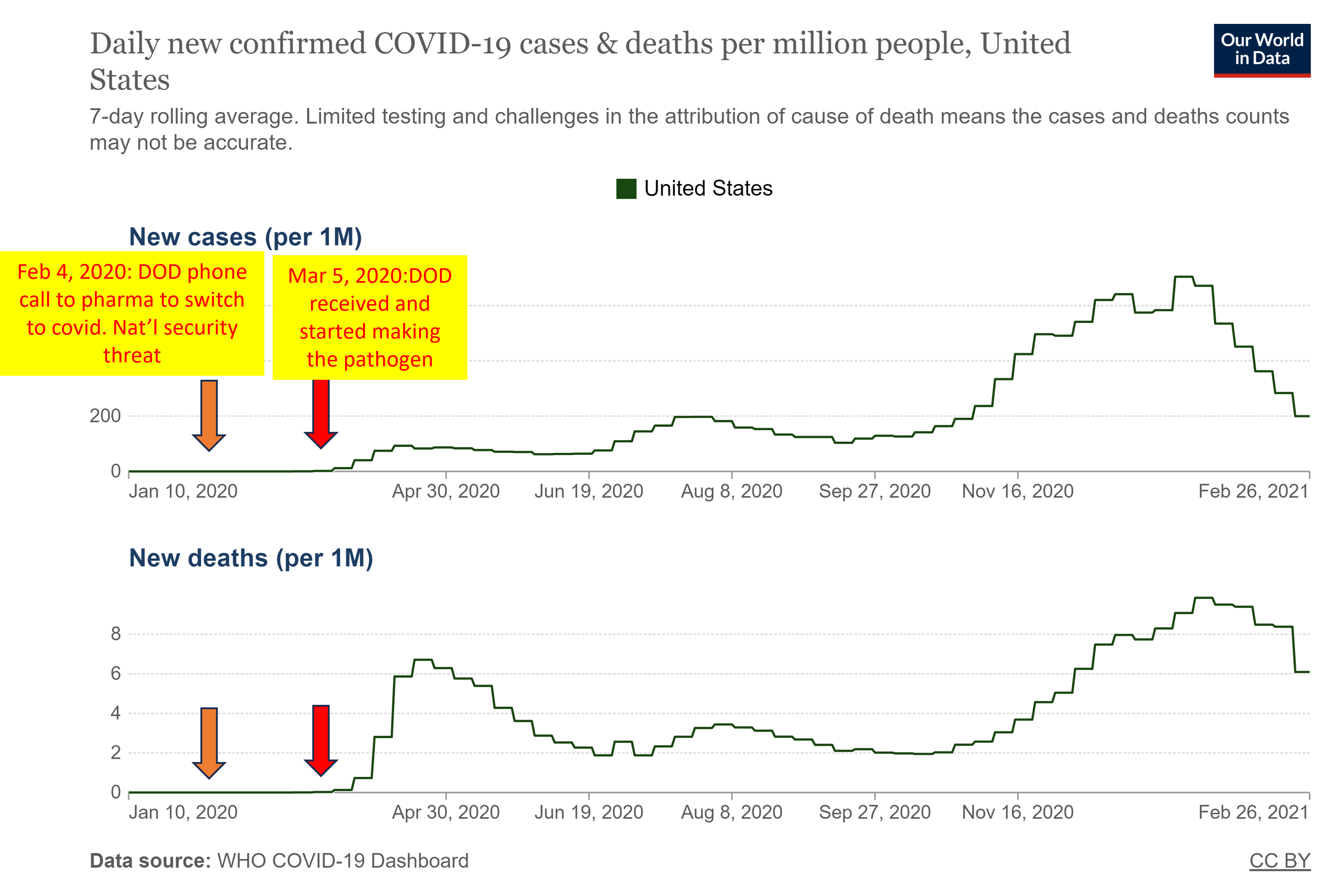
A leaked audio recording from an executive meeting at AstraZeneca from December 2020 became available[3]. The speakers included the CEO, Pascal Soirot and a Vice President responsible for the monoclonal antibody program, Mark Esser. Esser stated that the relationship between AstraZeneca and DOD started in 2017, when DOD approached the company with a proposal to rapidly (within 60 days!) develop countermeasures to novel viruses “identified” by the DOD. Esser stated that he had thought the idea was “more science fiction than science” at the time, but the company went along with it. Of course, the company wanted the DOD grants, and thus the pharmaceutical professionals reasonably questioning the unheard-of recklessness in product development were quickly silenced. Esser further stated that AstraZeneca and other members of the consortium received a phone call from the DOD on Feb 4, 2020, notifying them that covid was classified as a “national security threat” – a vague designation, with no hard definitions in law which is used by the military-intelligence apparatus to justify exceeding their authorities and violating real laws.
The military-pharma consortium that had made the investments into MCMs as early as 2017 was looking to cash in on their investments via a faked public crisis, a pandemic that had nothing to do with zoonotic viruses, but rather a false-flag attack with a non-lethal CBRN agent, followed by the government “public health measures” and “countermeasures” – mass unvalidated PCR testing, covid hospital murder protocols, lockdowns, then toxic shots.
At the same DOD press conference on March 5, 2020, Army Col. Wendy Sammons-Jackson, director of USAMRDC’s Military Infectious Disease Research Program stated that her group had received the pathogen and was growing and culturing the stock of it. At the time of this conference, there were practically no cases of covid in the US (fewer than 20) and no deaths. Why was the army making “more pathogen for stock”? This was justified to the public as necessary for making “countermeasures”, however the DOD was producing a much larger quantity of what they were calling the “virus” vs the actual virus or toxin that was circulating in the US – orders of magnitude more!


The cases and deaths began increasing after the DOD obtaining and making the stock of the pathogen, and after the lockdowns were announced on March 16, 2020. The “pandemic” was the result of the DOD and US Government activity, not a response to a natural event:
Hundreds of Covid countermeasures contracts became available via FOIA and SEC disclosures in redacted form.[1] These contracts allocated billions of dollars via DOD and Assistant Secretary of Pandemic Preparedness and Response at BARDA (Robert Kadlec under Trump Admin) to hundreds of companies – pharma, medical device, diagnostics, distribution, services and censorship/propaganda activities to maintain the fear, misery and isolation among the public so that they would view the covid vaccines as the only way back to “normal”.

These contracts specified the scope of deliverables as “demonstrations” and “prototypes” only. The contracts included the removal of all liability for the manufacturers and any contractors along the supply and distribution chain under the 2005 PREP Act and related federal legislation.
The contracts described covid countermeasures as intended for “civil and military application.”
The true nature of DOD’s involvement in the development and deployment of the injections was initially revealed publicly when Pfizer was forced to produce their DOD contract for “vaccine” in a motion to dismiss the whistleblower Brook Jackson v Ventavia case under False Claims Act in April 2022[1].
Pfizer’s motion boiled down to the plea of “we did not defraud the Government. We delivered the fraud that the Government ordered!” While morally abhorrent, the motion was legalistically correct: it was the Pentagon who ordered undisclosed “countermeasures” and “large scale manufacturing demonstrations” from pharma companies using the OTA/Defense Production contracting method. Besides numerous other problems with this, the word “demonstration” itself has an important meaning – it means a fake, something that is not real and is designed to fool the willing or unwilling target.
The Bait and Switch: Removal of the Pharmaceutical Consumer Safeguards for Covid Countermeasures
The officials lied about “FDA approved” status of Covid-19 injections. That lie enabled them to use the PREP Act liability shield since the actual distributed and injected substance was the Emergency Use (EUA) version of the product. At the same time, they were forcing vaccination mandates (impossible with experimental substances), by claiming that the two versions were “the same but legally distinct”. The details of the scam became available via the declaration by Peter Marks, Director of CBER at FDA. This declaration was filed in the court case where servicemembers were suing Secretary of Defense and the DOD for covid vaccination mandates.
Marks confirmed that the single criterion for EUA countermeasures was “maybe effective” decision of the HHS Secretary alone. He further stated that the FDA decided the EUA version must remain in circulation along with the “approved” version (which was never shipped in the US according to the shipping data made available by FOIA). This was an unprecedented move – EUA and fully licensed versions of the same product cannot not co-exist. This is because the key condition for an EUA is absence of an approved product for a severe, life threatening “emergency”. Yet, Pfizer had shipped billions of doses of Comirnaty outside of the US, and yet for some mysterious reason – not in the US. It was never “available”, except a tiny amount of 35,000 Comirnaty-[falsely]labeled doses that appeared in the US in August 2022 to maintain the pretense of availability.
Marks further explained that the BLA manufacturing sites have lot-release requirements, while the EUA ones do not. The lot release requirement is critical for assuring product quality, purity, stability, conformity to the label and other critical consumer safety parameters. Then he asserted that both BLA and EUA sites must adhere to the cGMP requirements but forgot to mention that for EUA manufacturing there is no way to verify or enforce the cGMP requirement. No enforcement means there is no real requirement.
Marks’s next statement was a breathtaking somersault - on the basis that maybe the same manufacturing facilities are used to make the non-existent-in-US BLA product (Comirnaty) as well as the EUA version, the FDA decided that lying to the people by withholding the information that the product they are receiving is EUA and thus cannot be forced on them was totally ok. This was communicated to all vaccinators in the United States. In this stunningly deceitful move, the FDA re-wrote the US law, exercising an authority that it never possessed. It is not legal for a pharmaceutical manufacturer to ship a drug labeled XYZ from the facilities that are authorized by the FDA to produce ABC and not XYZ. Simply claiming that they are “the same” does not solve this issue. Yet, the head of the Biologics division has instructed the healthcare providers to participate in this lie.
The covid “pandemic” and the “response”– the “public health” measures and the Operation Warp Speed – all of it was theater: lethal falsehoods wrapped into a veneer of on-paper-legal activities designed to fool the public into believing that the usurpation of government by “science experts” and removal of the human rights was justified by the Public Health Emergency of International Concern (PHEIC, pronounced “fake”), and that the poisonous weaponized bio-chemical injections were “safe and effective vaccines”. The injections marketed as vaccines were bioagents deployed by actors within the US Government and pharmaceutical/bioweapons industry, intended to injure and kill American people as targets, and exported to other countries' governments to injure and kill their people. All while reaping extraordinary profits and power from DOD/USG financial stakes in the MCM industry, and subversion of the Constitutional lawful governance and separation of powers under contrived, faked, state of emergency.
[1] https://www.iambrookjackson.com/_files/ugd/9df0bc_b7e94cf398e74b35a9182f27e685348b.pdf
[1] https://www.keionline.org/covid-contracts
[1] https://brownstone.org/articles/covid-mrna-vaccines-required-no-safety-oversight-part-two/
[2] https://www.keionline.org/bn-2020-3

[2] See, e.g., 21 U.S.C. § 355 (drugs); 42 U.S.C. § 262 (biologics).
[3] CFR Title 21, including sections in parts 1-99, 200-299, 300-499, 600-799, and 800-1299.
[4] Section 564 FD&C Act. Note that the EUA pathway should not be confused with the “Expanded Access Use” regulatory pathway which is often colloquially referred to as an “emergency use”. The expanded access is an investigational pathway and is regulated in the same manner as all normal drug approvals. (21 CFR 312.310-320)
[5] 21 U.S.C. § 360bbb-3(a)(1), (b).
[6] 21 USC 360bbb-3(k): If a product is the subject of an authorization under this section, the use of such product within the scope of the authorization shall not be considered to constitute a clinical investigation for purposes of section 355(i), 360b(j), or 360j(g) of this title or any other provision of this chapter or section 351 of the Public Health Service Act [42 U.S.C. 262].
[7] 21 U.S.C. § 360bbb-3(c)(2)(A)
[8] 21 U.S.C. § 360bbb3(c)(2)(B)
[9] 21 U.S.C. § 355(d)(2); See also 42 U.S.C. § 262(a)(2)(RB) (biologic approved only if it actually “is . . . safe”).
[10] https://www.fda.gov/media/143982/download
[11] 21 U.S.C. § 360bbb-3(e)(1)(A)(II) 21 USC 360bbb-3a: Emergency use of medical products (house.gov)
[12] 21 USC 360bbb-3a(c)
[13] https://www.govinfo.gov/content/pkg/PLAW-109publ417/pdf/PLAW-109publ417.pdf
[14] https://www.in.gov/dhs/files/FEMA-Fact-Sheet-COVID-Response-3.4.20.pdf
[15] OWS has been organizationally moved to BARDA under the name of “Next Gen”.
[16] See “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf” in Attachment
[1] https://virologyj.biomedcentral.com/articles/10.1186/1743-422X-2-69
[2] https://www.theguardian.com/world/2020/jun/10/surgisphere-sapan-desai-lancet-study-hydroxychloroquine-mass-audit-scientific-papers
[3] https://vaers.hhs.gov/about.html
[4] https://www.fda.gov/drugs/pharmaceutical-quality-resources/current-good-manufacturing-practice-cgmp-regulations
[5] www.howbad.info
[6] https://www.bitchute.com/channel/7dNrFbLeGSev/
[7] https://biotech.law.lsu.edu/blaw/fda/fdcact5a.htm
[8] https://www.bmj.com/content/372/bmj.n627
[9] https://phmpt.org/wp-content/uploads/2021/11/5.3.6-postmarketing-experience.pdf

[11] https://en.wikipedia.org/wiki/2008_Chinese_heparin_adulteration
Art for today: Bouquet of Roses, oil on panel, 11x14 in.
Sasha, even if your chapter is never 'published' I have retained it in my archives offline in an EMP-proof pouch with all my other valuable electronic documents from this era.
If we are lucky, our grandchildren will pull this from the wreckage of a smouldering crater and understand what happened.
God Bless you.
(I even retain all the references in full).
This is the most informative and concise summary of the COVID PsyOp I have read so far. Thank you so much (and Katherine) for your tireless work exposing this scam. The term "Psychopath" does not begin to describe those who pushed this evil program.